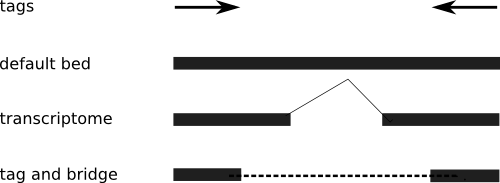
Multi-match reads in ChIP-Seq
I had an interesting comment left on my blog today, which is worth taking a few minutes to write a response to:
The first question asks about multi-matching locations - and if the point in question (point 3) applies to the Illumina Pipeline 1.3. Since point 3 was just that the older pipeline didn't provide the locations of the multi-matche reads, I suppose this no longer really applies: I understand the new version of Eland does provide multi-match alignment information, as do other aligners such as Bowtie. However, I should also mention that since I adopted Maq as my preferred aligner, I haven't used Eland much - so it's hard for me to give an informed opinion on the quality of the matches. I simply don't know if they're any good, and I won't belabour that point. I have used Bowtie specifically because it was able to do mutli-matches, but we didn't use it for ChIP-Seq, and the multi-matches had other uses in that experiment.
So, the more interesting question is whether I'd use multi-match reads in a ChIP-Seq analysis. And, off hand, my answer has to be no. But let me explain my reasoning, and the conditions in which I would change that answer.
First, lets assume that we have Single End Tags, so the multi-match information is not resolvable. That means anytime we have a read that maps to more than one location, we have the possibility that we can either map it to it's source - or we're mapping it incorrectly. A 50% change of "getting it right." The greater the number of multi-match locations, the smaller the chance we're actually finding it's correct origin. So, at best we've got a 50-50 chance that we're not adversely affecting the outcome of the experiment. That's not great.
In contrast, there are things we could do to make them usable. The most widely used method from FindPeaks is the weighted fragment distribution type. Thus, we could expand the principle to weight the fragments according to the number of sites. That would be... bearable. But would it significantly add to the quality of the alignment?
I'm still going to say no. Fragments we see in ChIP-Seq experiments tend to fall within 200-300bp of the regions in which the transcription factor (or other sites) bind. Thus, even if we were concerned that a particular transcription factor binds primarily to the similar motif regions at two sites, there should be more than enough (unique) sequence around that site (which is usually <30-40bp in length) to which you'll still see fragments aligning. That should compensate for the loss of the multi-match fragments.
Even more importantly, as read lengths increase, the amount of non-unique sequence decreases rapidly, making the shrinking number of multi-match reads less important.
The same argument can be extended for paired end tags: Just as read lengths improve and reduce the number of multi-match sites, more of the multi-match reads will be resolved by pairing them with a second read, which is unlikely to be within the same repeat region, thus reducing the number of reads that become unresolvable multi-matches. Proportionally, one would then expect that leaving out these reads become a smaller and smaller segment of the population, and would have to worry less and less about their contribution.
So, then, when would I want them?
Well, on the odd chance you're working with very short reads, you can pull off the weighting properly, and you have single end tags - and the multi-match reads make up a significant proportion of the reads, then it's worth exploring.
You'd need to start asking the tough questions: did the aligner simply find that a small k-mer of the read aligned to multiple locations (and was then unable to resolve the tie by extension the way some Eland aligners work)? Does the aligner use quality scores to identify mis-alignments? How reliable are the alignments (what's their error rate)? What was your sample, and how divergent is it from reference ? (e.g., cancer samples have a high variation rate, and so encourage many false alignments, making the alignments less reliable.)
Overall, I really don't see too many cases where you're going to gain a lot by digging in the multi-match files. That's not too say that you won't find anything good in there - you probably would, if you knew where to look, but the noise to signal ratio is going to be pretty poor - just by definition of the fact that they're mutli-match reads alone. You'll just have to ask if it's worth your time.
For the moment, I don't think my time (even at grad student wages) is worth it. It's just not low hanging fruit, when it comes to ChIP-Seq.
"Hi Anthony, I just discovered your blog and it looks very interesting to me!
Since this article on Eland is now more than one year old, I was wondering
if the description at point 3 about multi matching locations is still
applicable to the Eland program in the Illumina pipeline 1.3. More in general,
would you trust the multi matching locations extracted from the multi_eland
output files to perform a repeat enrichment analysis over an experiment of
ChIP-seq? If no, why? Thank you in advance for your attention."
The first question asks about multi-matching locations - and if the point in question (point 3) applies to the Illumina Pipeline 1.3. Since point 3 was just that the older pipeline didn't provide the locations of the multi-matche reads, I suppose this no longer really applies: I understand the new version of Eland does provide multi-match alignment information, as do other aligners such as Bowtie. However, I should also mention that since I adopted Maq as my preferred aligner, I haven't used Eland much - so it's hard for me to give an informed opinion on the quality of the matches. I simply don't know if they're any good, and I won't belabour that point. I have used Bowtie specifically because it was able to do mutli-matches, but we didn't use it for ChIP-Seq, and the multi-matches had other uses in that experiment.
So, the more interesting question is whether I'd use multi-match reads in a ChIP-Seq analysis. And, off hand, my answer has to be no. But let me explain my reasoning, and the conditions in which I would change that answer.
First, lets assume that we have Single End Tags, so the multi-match information is not resolvable. That means anytime we have a read that maps to more than one location, we have the possibility that we can either map it to it's source - or we're mapping it incorrectly. A 50% change of "getting it right." The greater the number of multi-match locations, the smaller the chance we're actually finding it's correct origin. So, at best we've got a 50-50 chance that we're not adversely affecting the outcome of the experiment. That's not great.
In contrast, there are things we could do to make them usable. The most widely used method from FindPeaks is the weighted fragment distribution type. Thus, we could expand the principle to weight the fragments according to the number of sites. That would be... bearable. But would it significantly add to the quality of the alignment?
I'm still going to say no. Fragments we see in ChIP-Seq experiments tend to fall within 200-300bp of the regions in which the transcription factor (or other sites) bind. Thus, even if we were concerned that a particular transcription factor binds primarily to the similar motif regions at two sites, there should be more than enough (unique) sequence around that site (which is usually <30-40bp in length) to which you'll still see fragments aligning. That should compensate for the loss of the multi-match fragments.
Even more importantly, as read lengths increase, the amount of non-unique sequence decreases rapidly, making the shrinking number of multi-match reads less important.
The same argument can be extended for paired end tags: Just as read lengths improve and reduce the number of multi-match sites, more of the multi-match reads will be resolved by pairing them with a second read, which is unlikely to be within the same repeat region, thus reducing the number of reads that become unresolvable multi-matches. Proportionally, one would then expect that leaving out these reads become a smaller and smaller segment of the population, and would have to worry less and less about their contribution.
So, then, when would I want them?
Well, on the odd chance you're working with very short reads, you can pull off the weighting properly, and you have single end tags - and the multi-match reads make up a significant proportion of the reads, then it's worth exploring.
You'd need to start asking the tough questions: did the aligner simply find that a small k-mer of the read aligned to multiple locations (and was then unable to resolve the tie by extension the way some Eland aligners work)? Does the aligner use quality scores to identify mis-alignments? How reliable are the alignments (what's their error rate)? What was your sample, and how divergent is it from reference ? (e.g., cancer samples have a high variation rate, and so encourage many false alignments, making the alignments less reliable.)
Overall, I really don't see too many cases where you're going to gain a lot by digging in the multi-match files. That's not too say that you won't find anything good in there - you probably would, if you knew where to look, but the noise to signal ratio is going to be pretty poor - just by definition of the fact that they're mutli-match reads alone. You'll just have to ask if it's worth your time.
For the moment, I don't think my time (even at grad student wages) is worth it. It's just not low hanging fruit, when it comes to ChIP-Seq.
Labels: Aligners, Bioinformatics, biology, Chip-Seq, Eland, FindPeaks, paired end tags, Solexa/Illumina